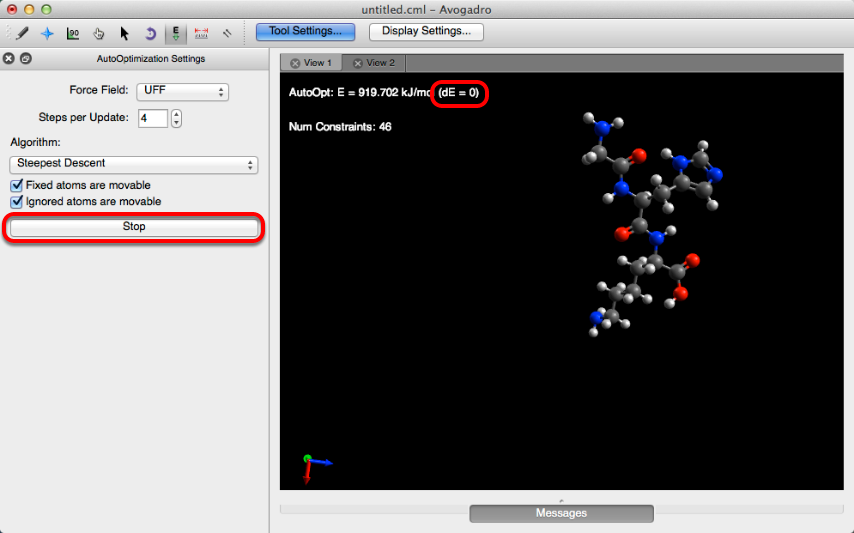
Auto Optimize Tool
The Auto Optimize tool continuously optimizes molecular geometry through molecular mechanics. This tool provides an interactive interface, allowing you to manipulate a molecule while it’s molecular geometry is being optimized.
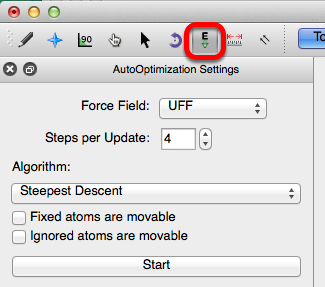
Force Fields & Algorithms
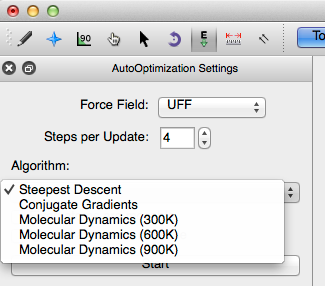
The Auto-Optimization settings provide several force field options. The default force field in Avogadro is UFF (Universal Force Field). UFF can generally reproduce the most structural features across the periodic table. However, depending on the molecule being optimized, the other force fields may be better suited to optimize the molecular parameters. The force field options are shown below. For more information on force fields refer to the optimizing geometry section of this lab manual.
The default setting for “Steps per Update” is 4. This number can be preferentially increased or decreased. If you have a slower computer consider decreasing this number.
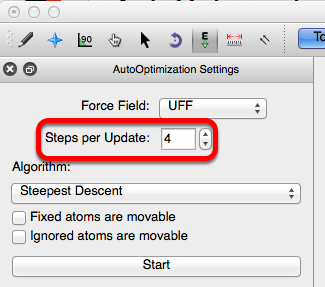
Avogadro also has the ability to apply specific algorithms dependent on your need. Steepest descent is the default algorithm, and has the most fluid and interactive system.
Fixing & Ignoring Selections
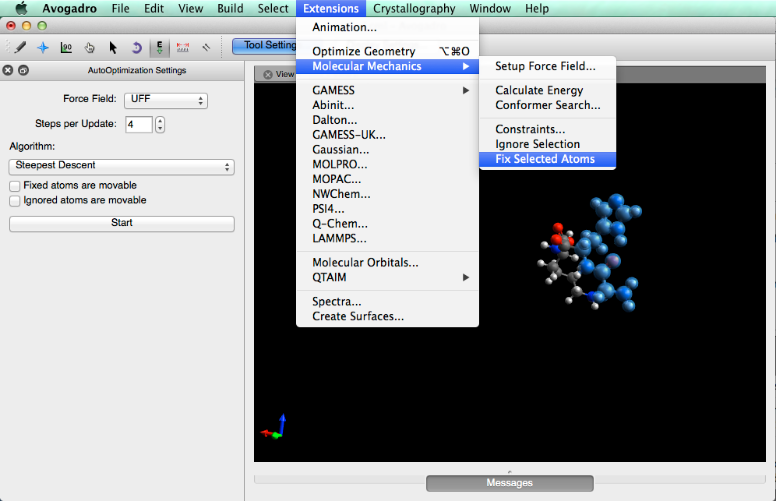
If necessary, atoms can be fixed into place before optimization so that they don’t move. This is done by going to the “Extensions” menu, holding your cursor over “Molecular Mechanics” and then selecting “Fix Selected Atoms”.
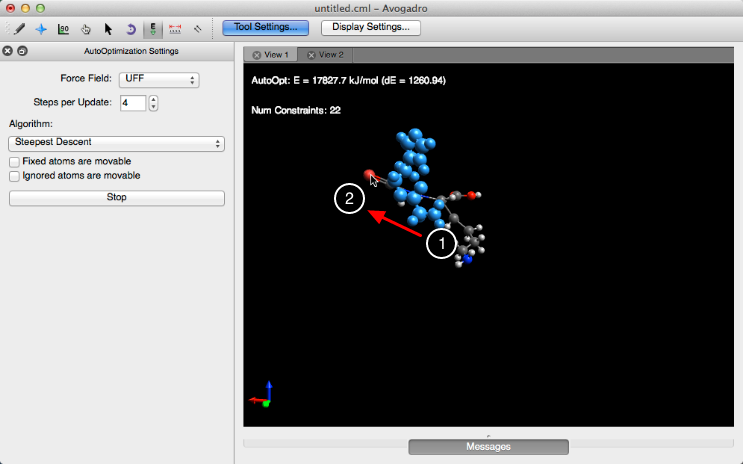
Clicking on “Start” will allow you to manipulate the molecule by left clicking on an atom and dragging your cursor. Notice that the fixed atoms don’t bend with the manipulation of the molecule.
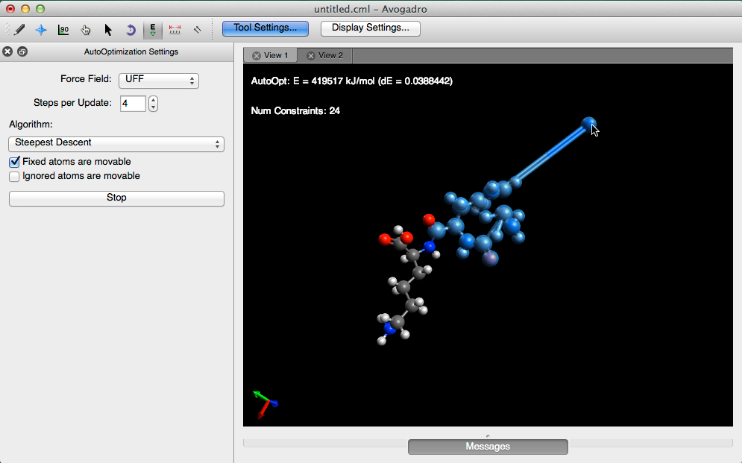
If the “Fixed atoms are movable” box is checked, the bonds between atoms can be manipulated into a new configuration. Manipulating a bond using this feature will lock that bond into place, and display the number of constraints cast on that molecule. Below is an exaggerated version of a bond manipulation using this feature.
Selecting the “Extensions” drop down menu, holding your cursor over the “Molecular Mechanics” option, and choosing “Ignore Selection” will allow you to ignore a selection of atoms. Avogadro registers an ignored selection as if the selection doesn’t exist. Therefore optimization only takes place in correspondence to atoms that aren’t ignored. Atoms that have been ignored can still be adjusted if the “Ignored atoms are movable” box is checked, however they will not be optimized.
Molecules will reoptimize until dE=0 or “Stop” is clicked.