Constraints & Optimizations
Avogadro allows for the optimization of an object, with respect to a variable(s). Below is one example of how constraints can be applied while optimizing a molecule.
Constraints
Constraints can be applied to fix or ignore a specific selection of atoms in a molecule, as well as to fix distances, and angles. For additional information review the “Auto-Optimization Tool” and “The Extensions Menu” sections of this manual.
Example
Let’s say we have a diene, that we want to fix in a cisoid conformation before optimizing the geometry of the rest of the molecule (example shown below). After drawing your molecule, check the label display type, and click the wrench to the right of the name. Select the “Atom number” label form and close the dialog box.
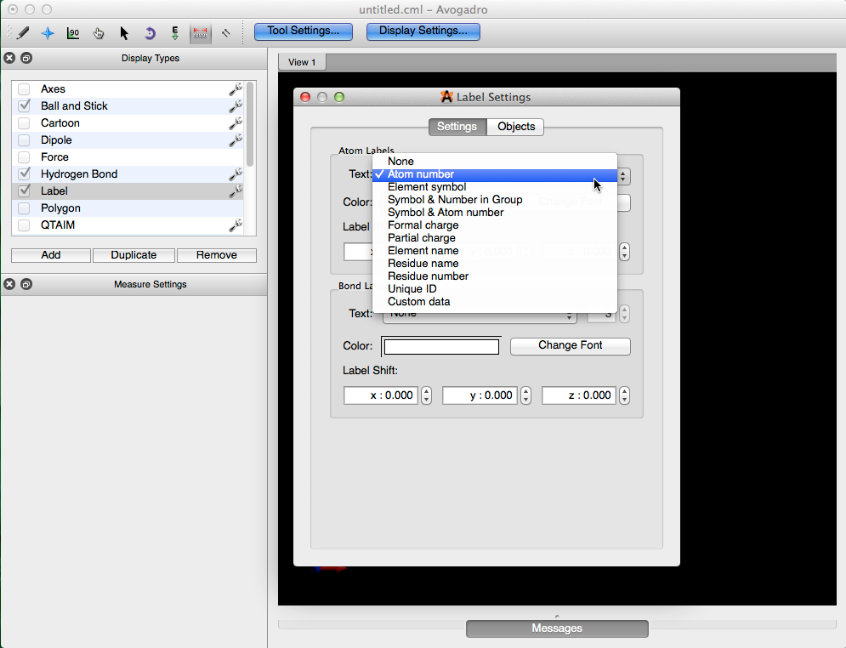
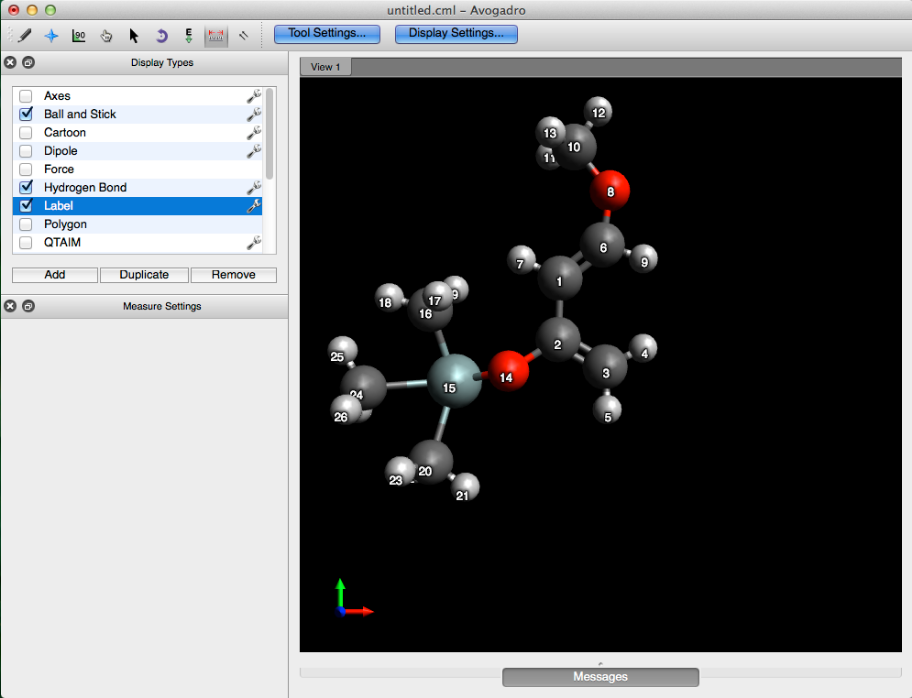
This will label all of the atom indices.
Before we can apply the constraint, we’ll need to figure out the distance between atom 3 & 6. Following the procedure displayed below, select the measure tool, and then choose the two atoms that you want to apply a constraint to. This will output a distance in angstroms at the bottom of the screen.
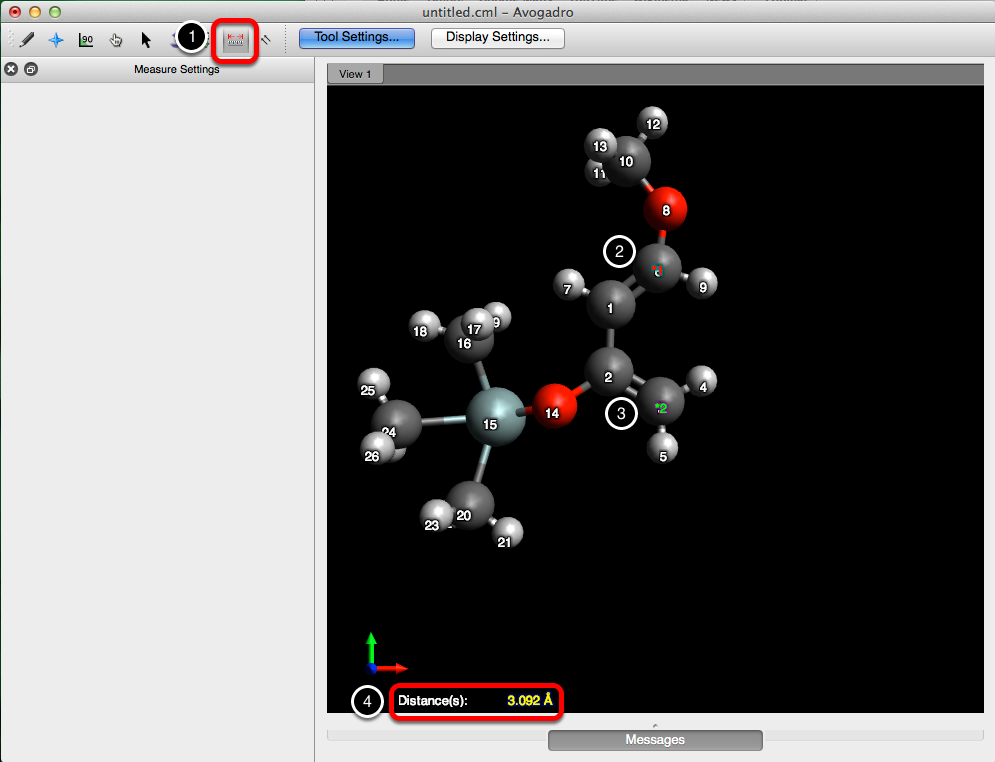
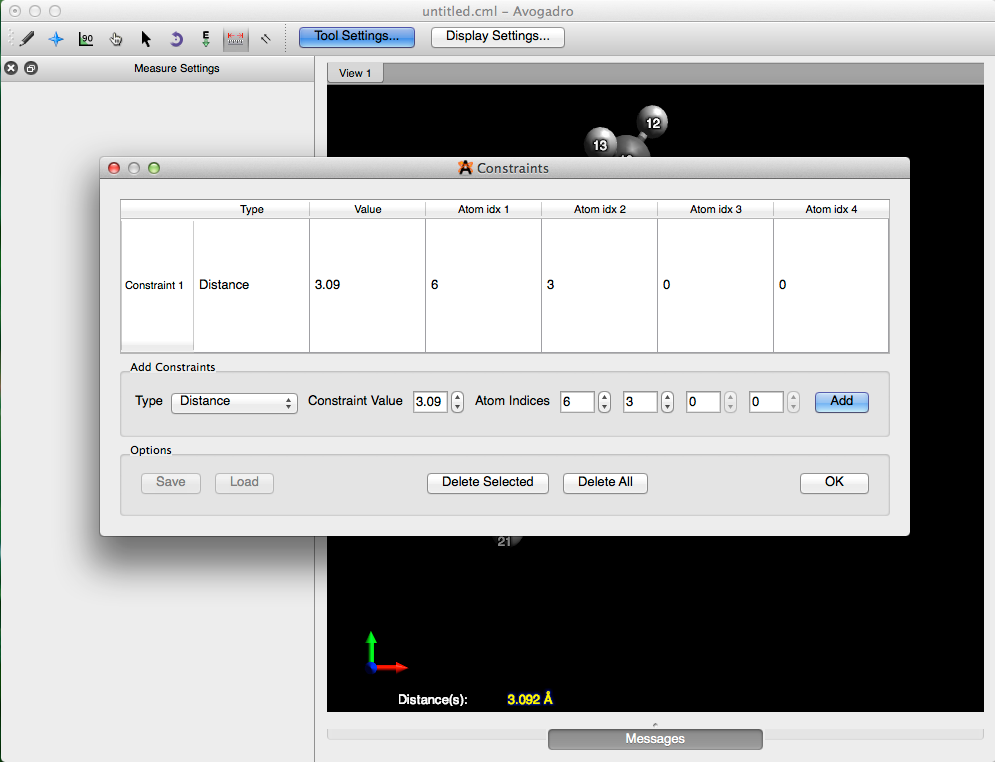
From here go to the “Extensions” menu, and under “Molecular Mechanics”, select “Constraints…”. Then choose “Distance”, as the type of constraint, enter in the length (3.092 Å), and the atom indices. Select “Add”, and then click “OK”, to add the constraint and close the dialog box.
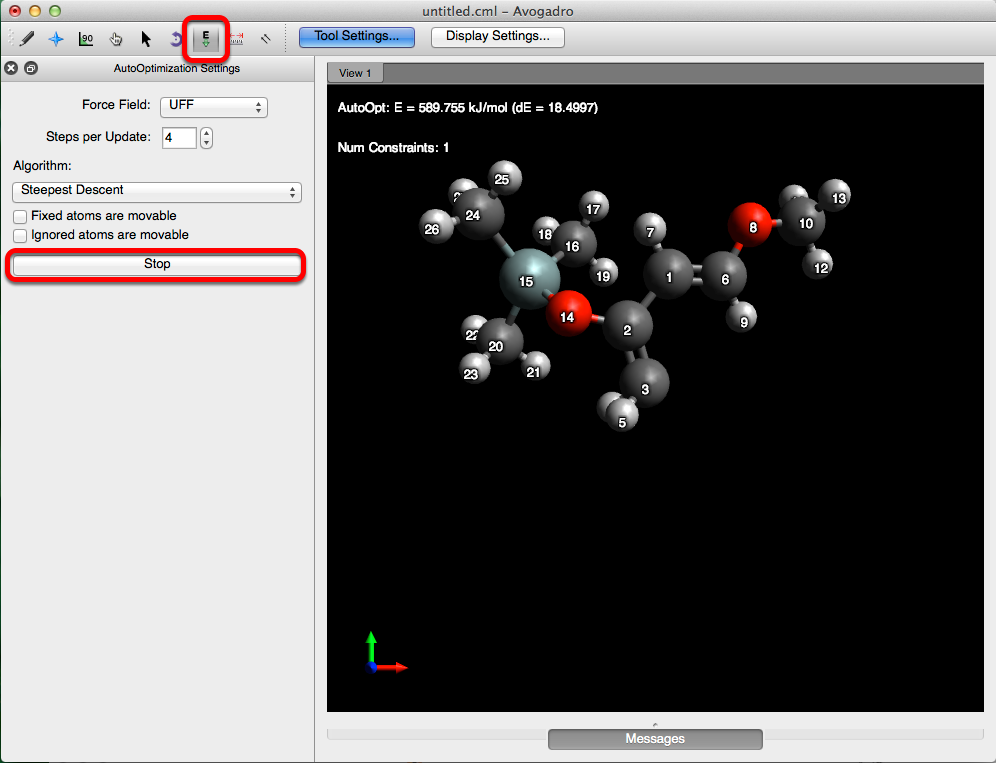
Optimizations
Optimizations can then be applied to work around the constraint. For more information about force fields refer to the “Molecular Mechanics and Force Fields” section of this manual.
Example cont.
Now (post addition of constraint), Avogadro will selectively keep the cisoid conformation while concurrently adjusting the parameters of the other atoms in the molecule.