LAMMPS input for water
Prepare initial geometry
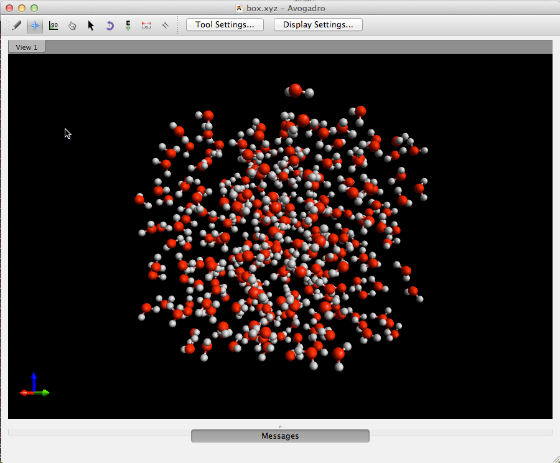
The independently developed Packmol extension can be used to generate a box of water molecules.
Open the LAMMPS input dialog
Prepare simulation parameters
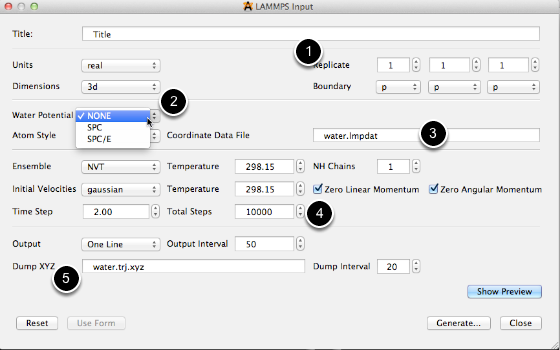
- Choose the number of repeating units of the input coordintes in x, y and z directions
- Choose the water potential. The current version supports SPC and SPC/E model potentials
- Choose the name of LAMMPS formatted coordinates. The name will be used in a later step when the lmpdat file is created.
- Choose the total number of MD steps.
- Choose the file name of the XYZ formatted trajectory file.
Generate the LAMMPS parameters file
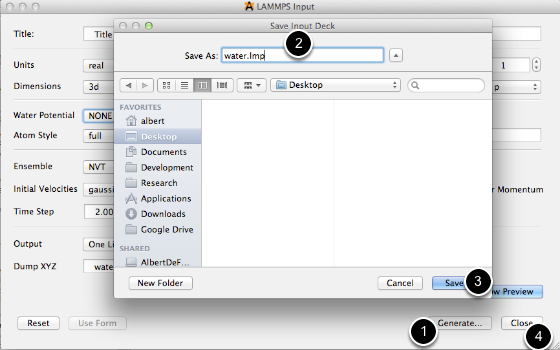
- Click the Generate button
- Choose a file name
- Click save
- Close the input generator dialog
Generate the LAMMPS Coordintes file
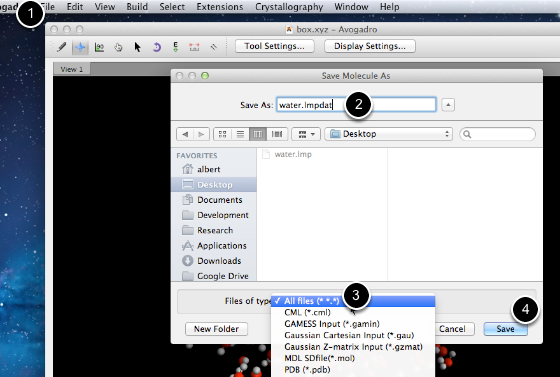
- Select “Save As” from the file menu
- Input the “water.lmpdat” file name from above
- Select “All files”
- Save the LAMMPS formatted coordinates file
Run LAMMPS
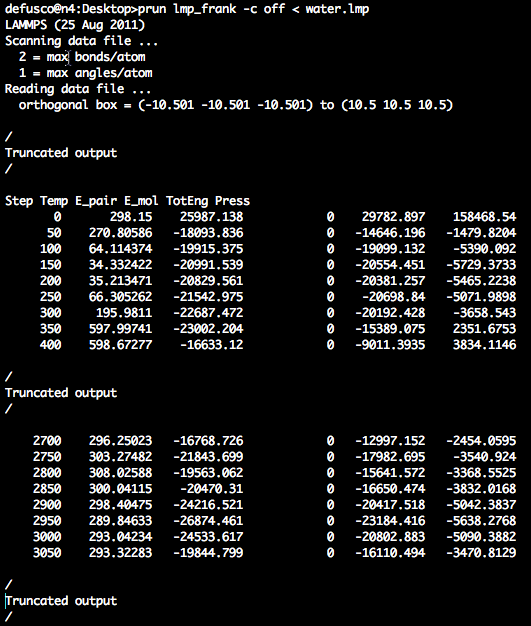
After 2700 time steps, the temperature is begining to stabilize.